学習院大学 理学部 化学科 / 自然科学研究科 化学専攻 岩田研究室
岩田研究室
<多様化・複雑化した試料を対象とした動的構造の分子レベル解明に向けて>
現代の基礎物理化学が扱う対象は,基本的な分子から,生きた細胞・ナノ構造体・機能性高分子材料まで多岐にわたる.ラマン分光法は,これらの多種多様な試料がもつ分子構造を観測するうえで,最も基本的な手法のひとつである.ラマン分光法では試料の形態を選ばず,特別な前処理の必要がない.また,時間分解分光・顕微分光といった,他の分光手法との組み合わせが容易に可能である.一方,次のような問題点を持つ.
- 試料が蛍光を発することが多い.蛍光により測定が妨害される.
- 大きな光子エネルギーにより,試料を傷めやすい.
<現在の近赤外ラマン分光法の特徴と問題点>
現在用いられている近赤外ラマン分光法は,主に通常の自発ラマン分光法と,フーリエ変換ラマン分光法の2つである.これらは以下のような特徴および問題点を持つ.
- 自発ラマン分光法では,感度の良さは検出器の感度に大きく依存することになる.近年,現在最高の性能をもつ近赤外検出器(量子効率4 %;可視光検出器の10 %程度)を用いて,明瞭な近赤外ラマンスペクトルが報告されている.しかしながら,現時点でこれと同等の検出器を入手することはきわめて困難である.
- フーリエ変換近赤外ラマン分光法では,干渉計とフーリエ変換の手法を組み合わせ,検出器の雑音の大きさを克服している.しかし,検出器の感度が低いうえ,干渉光学系によってラマン散乱光を大きく損失するため,ラマン散乱光の検出効率は低い.
<近赤外誘導ラマン分光法>
そこで,われわれは近赤外領域の2本の光(ラマン励起光:波長1190 nm,ラマンプローブ光:波長900~1600 nmの白色光)を試料に照射し,非線形ラマン効果のひとつ「誘導ラマン散乱」のスペクトルを観測することにした.誘導ラマン散乱は,ラマン活性な振動が多数の分子について一斉に励振されることにより,ラマン励起光から見てちょうどストークスラマン散乱あるいはアンチストークスラマン散乱と同じ波長を持つラマンプローブ光の強度が変化する現象である(図1).そのため,誘導ラマン散乱はラマン散乱と同じ振動情報を与える.誘導ラマンスペクトルを記録するにはラマンプローブ光のわずかな強度変化を検出する必要があるが,これに対してはわれわれがこれまでに蓄積してきた近赤外吸収分光の技術を応用することができる.したがって,近赤外誘導ラマンスペクトルを多波長で同時測定できるマルチプレックス分光計は,近赤外ラマン分光の有力な手法のひとつとなる.
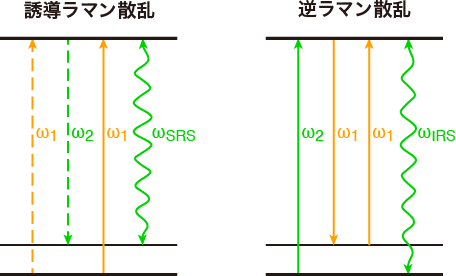
図1 (左)誘導ラマン散乱のエネルギー図.ストークスラマン散乱と同じ振動数の光の強度が増大する.(右)逆ラマン散乱のエネルギー図.アンチストークスラマン散乱と同じ振動数の光の強度が減少する.
<フェムト秒時間分解マルチプレックス近赤外誘導ラマン分光計>
われわれが製作したフェムト秒時間分解マルチプレックス近赤外誘導ラマン分光計のブロック図を図2に示す.モード同期チタンサファイアレーザーの再生増幅パルス出力(800 nm, 3.5 mJ, 1 kHz)を3つに分ける.1つめの出力を光パラメトリック増幅器に導入し,波長250~800 nmで可変なポンプ光パルスを得る.2つめの出力を別の光パラメトリック増幅器によって波長1190 nmの近赤外光に変換し,反射型狭帯域バンドパスフィルターを用いて波数幅約3 cm-1のラマンポンプ光パルスを得る.3つめの出力をサファイア板に緩く絞り,波長900~1550 nmにかけて広がるプローブ光パルスを得る.ポンプ光パルスによって試料を光励起し,次いでラマンポンプ光パルスとプローブ光パルスを同時に照射して誘導ラマン散乱を発生させる.試料を通過したプローブ光パルスの強度を,分光器とInGaAs検出器(512素子)を用いて512波長について同時検出する.ラマンポンプ光パルスの照射/非照射を光チョッパーで100 msごとに切り替え,誘導ラマン散乱によるプローブ光パルスの強度変化を感度良く検出し,誘導ラマンスペクトルを得る.ポンプ光パルスとラマンポンプ光・プローブ光パルスの間の時間遅延を光学遅延回路で制御し,時間分解スペクトルを測定する.光学遅延回路・分光器・検出器は自作のプログラムで自動制御される.また,光チョッパーの回転に同期して検出器の信号取り込みを行うため,自作の簡単な電子回路を用いている.本分光計の時間応答関数の半値全幅は240 fsであり,約5 cm-1までの波数分解が可能である.
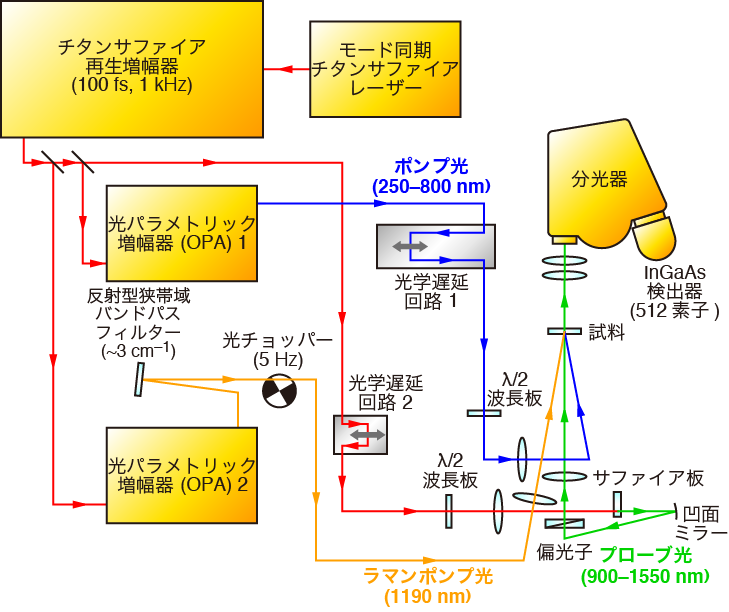
図2 フェムト秒時間分解マルチプレックス近赤外誘導ラマン分光計のブロック図.
<β-カロテンS2状態の誘導ラマンスペクトル測定>
β-カロテンは生体内に存在する代表的な分子のひとつである.β-カロテンは直鎖状の共役ポリエン構造を持ち(図3),光吸収およびクロロフィルへのエネルギー移動などの役割を担っていると考えられている.励起状態にあるβ-カロテンの分子構造,電子構造,緩和動力学を観測することで,β-カロテンをはじめとするカロテノイドが生体内において担う役割を理解するうえで重要な知見が得られる.
図3 β-カロテンの構造式.
これまでに,β-カロテンは可視光を吸収してS2状態に遷移し,約200 fsの時定数でS1状態へと内部転換することが報告されている(図4).ここでβ-カロテンS2状態のラマンスペクトルが得られれば,S2状態の共役ポリエン構造に関して直接かつ詳細な情報を与えるきわめて重要なデータとなる.しかし,S2状態のラマンスペクトルは未だほとんど報告されていない.その理由として,
- 近赤外共鳴ラマン分光の手法が必要であること
- フェムト秒オーダーの時間応答関数を持った分光計が必要であること
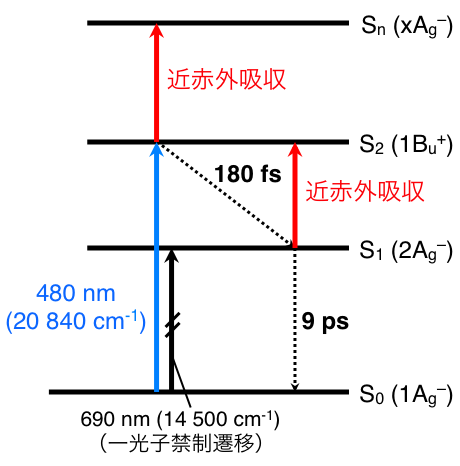
図4 β-カロテンの電子状態のエネルギー図.
われわれは,フェムト秒時間分解マルチプレックス近赤外誘導ラマン分光計を用いて,β-カロテンS2状態の誘導ラマンスペクトルを明瞭に観測した.波長480 nmの光を照射したところ,β-カロテンS2状態の同位相C=C伸縮振動による誘導ラマンバンドが1580 cm-1に強く観測された(図5).きわめて大きいバンド幅は,S2状態の寿命が短いことによる.また,同位相C-C伸縮振動およびCH3横揺れ振動による誘導ラマンバンドがそれぞれ1240, 1050 cm-1に観測された.一方,1776 cm-1の誘導ラマンバンドはS1状態の同位相C=C伸縮振動に帰属される.
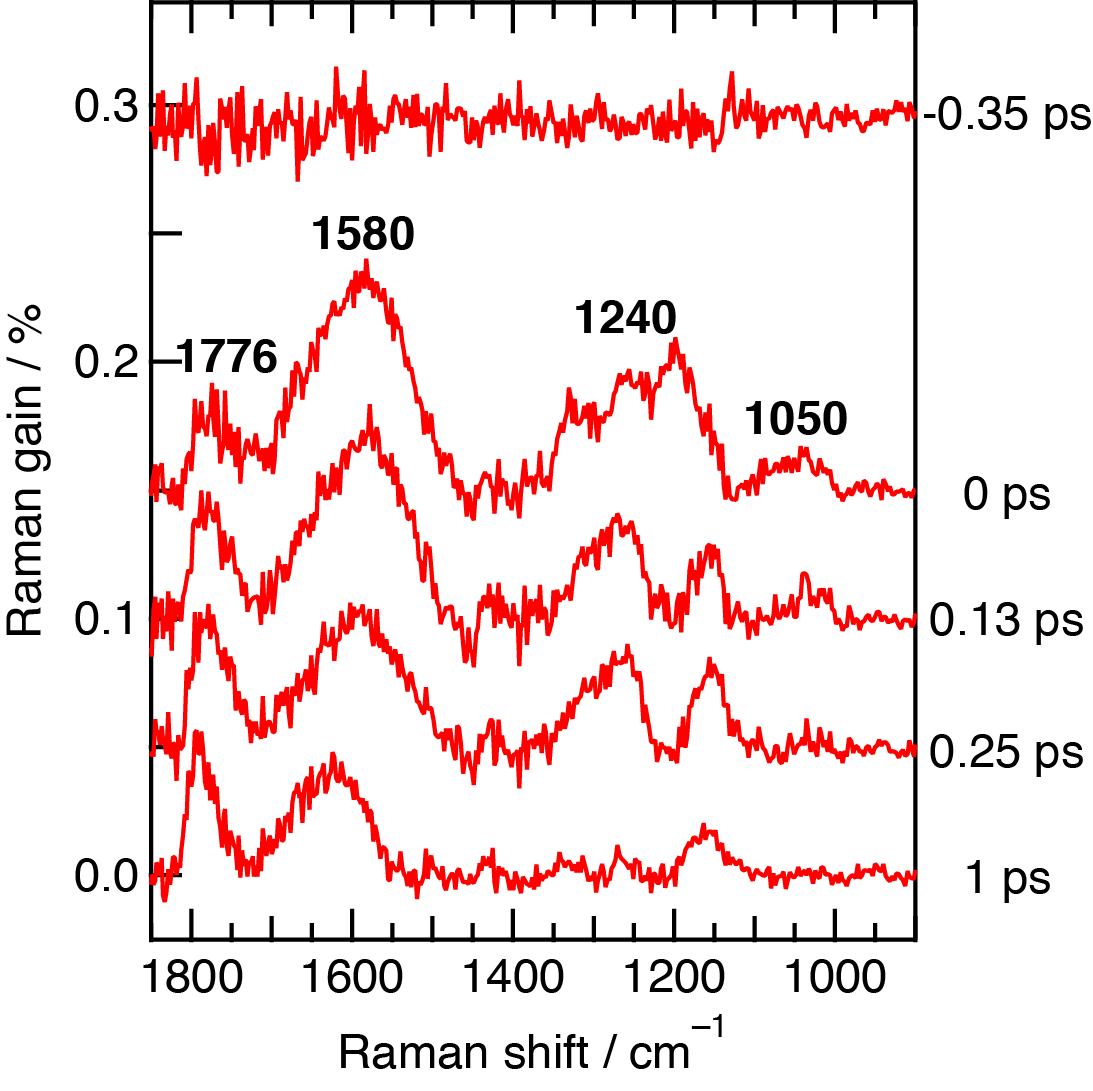
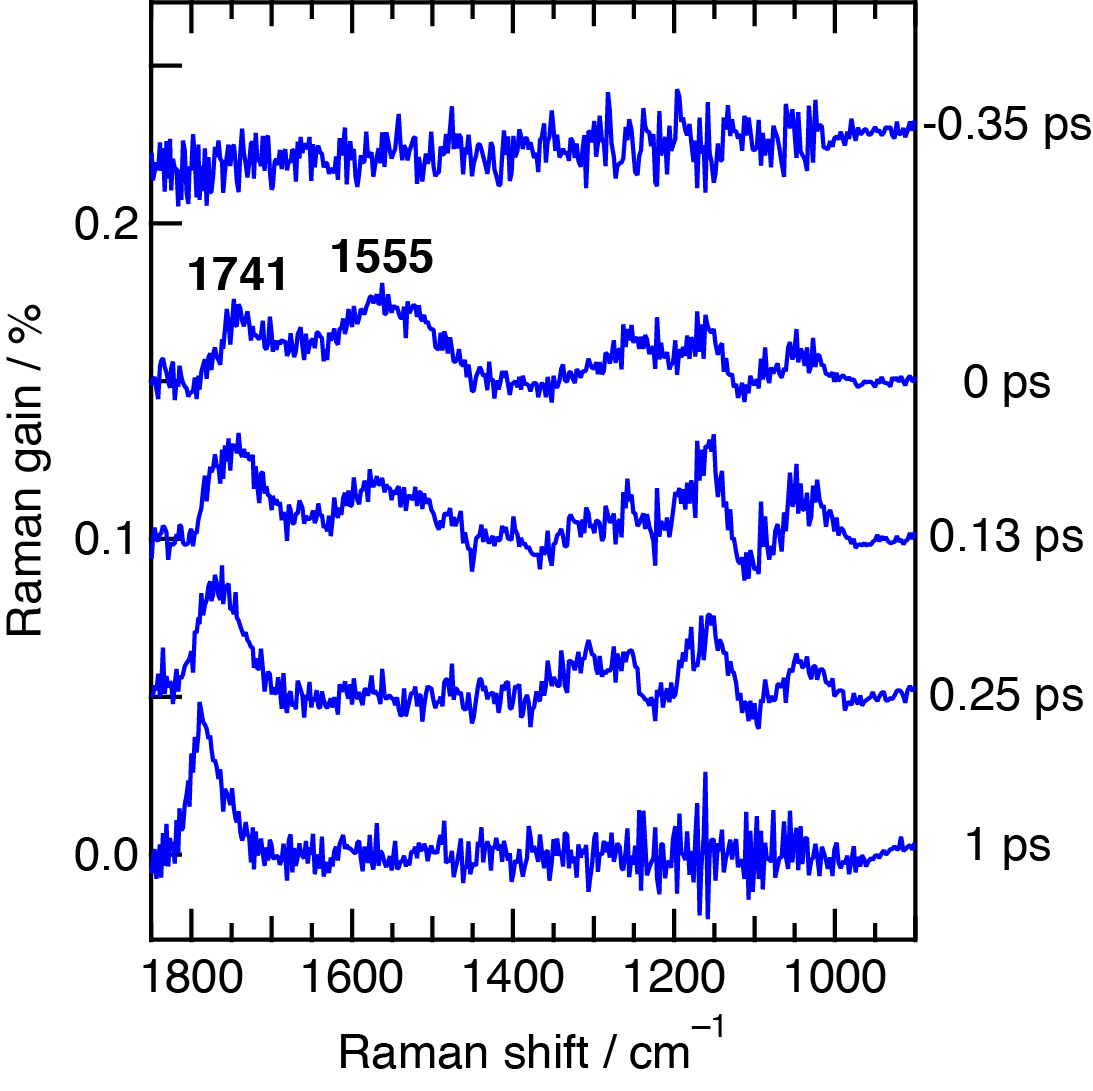
図5 (左)β-カロテンを波長480 nmの光で励起して測定したフェムト秒時間分解近赤外誘導ラマンスペクトル.(右)β-カロテンを波長403 nmの光で励起した場合の近赤外誘導ラマンスペクトル.
β-カロテンに波長403 nmの光を照射して得られたスペクトルでは,S2状態の同位相C=C伸縮振動が1555 cm-1に, S1状態の同位相C=C伸縮振動が1741 cm-1にそれぞれ観測された(図5).S2状態の同位相C=C伸縮振動のバンド位置が波長480 nmの光で励起した場合よりも低波数となったのは振動の非調和性に起因する.一方,S1状態の同位相C=C伸縮振動のバンド位置が低波数となったのは,S2状態が持っている余剰エネルギーがS2状態からS1状態への内部転換に影響を与えていることを示している.われわれは励起波長に対する誘導ラマンバンドの位置の違い,およびバンド位置の時間変化の違いを解析し,S2状態からS1状態への緩和がどのように進行するかを説明するモデルを提案した(図6).
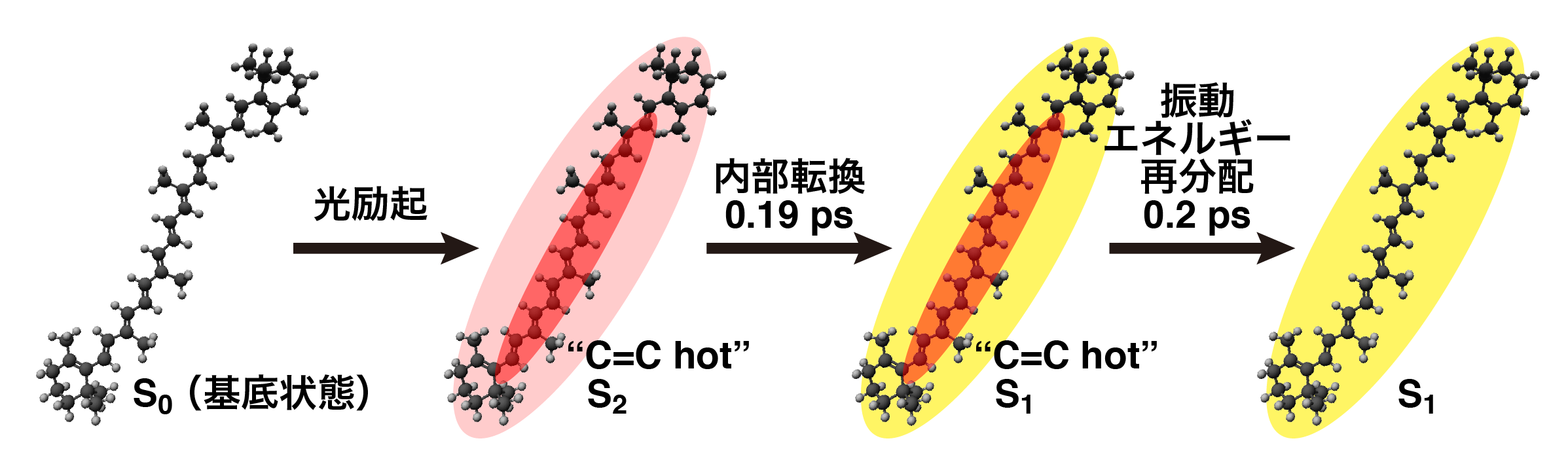
図6 β-カロテンのS2状態からS1状態への緩和の進行機構.
【参考文献】
[1] T. Takaya, K. Iwata, J. Phys. Chem. A, 2014, 118(23), 4071.